Analyzing Synonymous and Nonsynonymous Substitution Rates
This example shows how the analysis of synonymous and nonsynonymous mutations at the nucleotide level can suggest patterns of molecular adaptation in the genome of HIV-1. This example is based on the discussion of natural selection at the molecular level presented in Chapter 6 of "Introduction to Computational Genomics. A Case Studies Approach" [1].
Introduction
The human immunodeficiency virus 1 (HIV-1) is the more geographically widespread of the two viral strains that cause Acquired Immunodeficiency Syndrome (AIDS) in humans. Because the virus rapidly and constantly evolves, at the moment there is no cure nor vaccine against HIV infection. The HIV virus presents a very high mutation rate that allows it to evade the response of our immune system as well as the action of specific drugs. At the same time, however, the rapid evolution of HIV provides a powerful mechanism that reveals important insights into its function and resistance to drugs. By estimating the force of selective pressures (positive and purifying selections) across various regions of the viral genome, we can gain a general understanding of how the virus evolves. In particular, we can determine which genes evolve in response to the action of the targeted immune system and which genes are conserved because they are involved in some of the virus essential functions.
Nonsynonymous mutations to a DNA sequence cause a change in the translated amino acid sequence, whereas synonymous mutations do not. The comparison between the number of nonsynonymous mutations (dn or Ka), and the number of synonymous mutations (ds or Ks), can suggest whether, at the molecular level, natural selection is acting to promote the fixation of advantageous mutations (positive selection) or to remove deleterious mutations (purifying selection). In general, when positive selection dominates, the Ka/Ks ratio is greater than 1; in this case, diversity at the amino acid level is favored, likely due to the fitness advantage provided by the mutations. Conversely, when negative selection dominates, the Ka/Ks ratio is less than 1; in this case, most amino acid changes are deleterious and, therefore, are selected against. When the positive and negative selection forces balance each other, the Ka/Ks ratio is close to 1.
Extracting Sequence Information for Two HIV-1 Genomes
Download two genomic sequences of HIV-1 (GenBank® accession numbers AF033819 and M27323). For each encoded gene we extract relevant information, such as nucleotide sequence, translated sequence and gene product name.
hiv1(1) = getgenbank('AF033819'); hiv1(2) = getgenbank('M27323');
For your convenience, previously downloaded sequences are included in a MAT-file. Note that data in public repositories is frequently curated and updated; therefore the results of this example might be slightly different when you use up-to-date datasets.
load hiv1.mat
Extract the gene sequence information using the featureparse function.
genes1 = featureparse(hiv1(1),'feature','CDS','Sequence','true'); genes2 = featureparse(hiv1(2),'feature','CDS','Sequence','true');
Calculating the Ka/Ks Ratio for HIV-1 Genes
Align the corresponding protein sequences in the two HIV-1 genomes and use the resulting alignment as a guide to insert the appropriate gaps in the nucleotide sequences. Then calculate the Ka/Ks ratio for each individual gene and plot the results.
KaKs = zeros(1,numel(genes1)); for iCDS = 1:numel(genes1) % align aa sequences of corresponding genes [score,alignment] = nwalign(genes1(iCDS).translation,genes2(iCDS).translation); seq1 = seqinsertgaps(genes1(iCDS).Sequence,alignment(1,:)); seq2 = seqinsertgaps(genes2(iCDS).Sequence,alignment(3,:)); % Calculate synonymous and nonsynonymous substitution rates [dn,ds] = dnds(seq1,seq2); KaKs(iCDS) = dn/ds; end % plot Ka/Ks ratio for each gene bar(KaKs); ylabel('Ka / Ks') xlabel('genes') ax = gca; ax.XTickLabel = {genes1.product}; % plot dotted line at threshold 1 hold on line([0 numel(KaKs)+1],[1 1],'LineStyle', ':'); KaKs
KaKs =
Columns 1 through 7
0.2560 0.1359 0.3013 0.1128 1.1686 0.4179 0.5150
Columns 8 through 9
0.5115 0.3338

All the considered genes, with the exception of TAT, have a total Ka/Ks less than 1. This is in accordance with the fact that most protein-coding genes are considered to be under the effect of purifying selection. Indeed, the majority of observed mutations are synonymous and do not affect the integrity of the encoded proteins. As a result, the number of synonymous mutations generally exceeds the number of nonsynonymous mutations. The case of TAT represents a well known exception; at the amino acid level, the TAT protein is one of the least conserved among the viral proteins.
Calculating the Ka/Ks Ratio Using Sliding Windows
Oftentimes, different regions of a single gene can be exposed to different selective pressures. In these cases, calculating Ka/Ks over the entire length of the gene does not provide a detailed picture of the evolutionary constraints associated with the gene. For example, the total Ka/Ks associated with the ENV gene is 0.5155. However, the ENV gene encodes for the envelope glycoprotein GP160, which in turn is the precursor of two proteins: GP120 (residues 31-511 in AF033819) and GP41 (residues 512-856 in AF033819). GP120 is exposed on the surface of the viral envelope and performs the first step of HIV infection; GP41 is non-covalently bonded to GP120 and is involved in the second step of HIV infection. Thus, we can expect these two proteins to respond to different selective pressures, and a global analysis on the entire ENV gene can obscure diversified behavior. For this reason, we conduct a finer analysis by using sliding windows of different sizes.
Align ENV genes of the two genomes and measure the Ka/Ks ratio over sliding windows of size equal to 5, 45, and 200 codons.
env = 8; % ORF number corresponding to gene ENV % align the two ENV genes [score,alignment] = nwalign(genes1(env).translation,genes2(env).translation); env_1 = seqinsertgaps(genes1(env).Sequence,alignment(1,:)); env_2 = seqinsertgaps(genes2(env).Sequence,alignment(3,:)); % compute Ka/Ks using sliding windows of different sizes [dn1, ds1, vardn1, vards1] = dnds(env_1, env_2, 'window', 200); [dn2, ds2, vardn2, vards2] = dnds(env_1, env_2, 'window', 45); [dn3, ds3, vardn3, vards3] = dnds(env_1, env_2, 'window', 5); % plot the Ka/Ks trends for the different window sizes figure() hold on plot(dn1./ds1, 'r'); plot(dn2./ds2, 'b'); plot(dn3./ds3, 'g'); line([0 numel(dn3)],[1 1],'LineStyle',':'); legend('window size = 200', 'window size = 45', 'window size = 5'); ylim([0 10]) ylabel('Ka / Ks') xlabel('sliding window (starting codon)') title 'Env';

The choice of the sliding window size can be problematic: windows that are too long (in this example, 200 codons) average across long regions of a single gene, thus hiding segments where Ka/Ks is potentially behaving in a peculiar manner. Too short windows (in this example, 5 codons) are likely to produce results that are very noisy and therefore not very meaningful. In the case of the ENV gene, a sliding window of 45 codons seems to be appropriate. In the plot, although the general trend is below the threshold of 1, we observe several peaks over the threshold of 1. These regions appear to undergo positive selection that favors amino acid diversity, as it provides some fitness advantage.
Using Sliding Window Analyses for GAG, POL and ENV Genes
You can perform similar analyses on other genes that display a global Ka/Ks ratio less than 1. Compute the global Ka/Ks ratio for the GAG, POL and ENV genes. Then repeat the calculation using a sliding window.
gene_index = [1;2;8]; % ORF corresponding to the GAG, POL, ENV genes windowSize = 45; % display the global Ka/Ks for the GAG, POL and ENV genes KaKs(gene_index) for i = 1:numel(gene_index) ID = gene_index(i); [score,alignment] = nwalign(genes1(ID).translation,genes2(ID).translation); s1 = seqinsertgaps(genes1(ID).Sequence,alignment(1,:)); s2 = seqinsertgaps(genes2(ID).Sequence,alignment(3,:)); % plot Ka/Ks ratio obtained with the sliding window [dn, ds, vardn, vards] = dnds(s1, s2, 'window', windowSize); figure() plot(dn./ds, 'b') line([0 numel(dn)],[1 1], 'LineStyle', ':') ylabel('Ka / Ks') xlabel('sliding window (starting codon)') title(genes1(ID).product); end
ans =
0.2560 0.1359 0.5115



The GAG (Group-specific Antigen) gene provides the basic physical infrastructure of the virus. It codes for p24 (the viral capsid), p6 and p7 (the nucleocapsid proteins), and p17 (a matrix protein). Since this gene encodes for many fundamental proteins that are structurally important for the survival of the virus, the number of synonymous mutations exceeds the number of nonsynonymous mutations (i.e., Ka/Ks <1). Thus, this protein is expected to be constrained by purifying selection to maintain viral infectivity.
The POL gene codes for viral enzymes, such as reverse transcriptase, integrase, and protease. These enzymes are essential to the virus survival and, therefore, the selective pressure to preserve their function and structural integrity is quite high. Consequently, this gene appears to be under purifying selection and we observe Ka/Ks ratio values less than 1 for the majority of the gene length.
The ENV gene codes for the precursor to GP120 and GP41, proteins embedded in the viral envelope, which enable the virus to attach to and fuse with target cells. GP120 infects any target cell by binding to the CD4 receptor. As a consequence, GP120 has to maintain the mechanism of recognition of the host cell and at the same time avoid the detection by the immune system. These two roles are carried out by different parts of the protein, as shown by the trend in the Ka/Ks ratio. This viral protein is undergoing purifying (Ka/Ks < 1) and positive selection (Ka/Ks >1) in different regions. A similar trend is observed in GP41.
Analyzing the Ka/Ks Ratio and Epitopes in GP120
The glycoprotein GP120 binds to the CD4 receptor of any target cell, particularly the helper T-cell. This represents the first step of HIV infection and, therefore, GP120 was among the first proteins studied with the intent of finding a HIV vaccine. It is interesting to determine which regions of GP120 appear to undergo purifying selection, as indicators of protein regions that are functionally or structurally important for the virus survival, and could potentially represent drug targets.
From ENV genes, extract the sequences coding for GP120. Compute the Ka/Ks over sliding window of size equal to 45 codons. Plot and overlap the trend of Ka/Ks with the location of four T cell epitopes for GP120.
% GP120 protein boundaries in genome1 and genome2 respectively gp120_start = [31; 30]; % protein boundaries gp120_stop = [511; 501]; gp120_startnt = gp120_start*3-2; % nt boundaries gp120_stopnt = gp120_stop*3; % align GP120 proteins and insert appropriate gaps in nt sequence [score,alignment] = nwalign(genes1(env).translation(gp120_start(1):gp120_stop(1)), ... genes2(env).translation(gp120_start(2):gp120_stop(2))); gp120_1 = seqinsertgaps(genes1(env).Sequence(gp120_startnt(1):gp120_stopnt(1)),alignment(1,:)); gp120_2 = seqinsertgaps(genes2(env).Sequence(gp120_startnt(2):gp120_stopnt(2)),alignment(3,:)); % Compute and plot Ka/Ks ratio using the sliding window [dn120, ds120, vardn120, vards120] = dnds(gp120_1, gp120_2, 'window', windowSize); % Epitopes for GP120 identified by cellular methods (see reference [2]) epitopes = {'TVYYGVPVWK','HEDIISLWQSLKPCVKLTPL',... 'EVVIRSANFTNDAKATIIVQLNQSVEINCT','QIASKLREQFGNNK',... 'QSSGGDPEIVTHSFNCGGEFF','KQFINMWQEVGKAMYAPP',... 'DMRDNWRSELYKYKVVKIEPLGVAP'}; % Find location of the epitopes in the aligned sequences: epiLoc = zeros(numel(epitopes),2); for i = 1:numel(epitopes) [sco,ali,ind] = swalign(alignment(1,:),epitopes{i}); epiLoc(i,:) = ind(1) + [0 length(ali)-1]; end figure hold on % plot Ka/Ks relatively to the middle codon of the sliding window plot(windowSize/2+(1:numel(dn120)),dn120./ds120) plot(epiLoc,[1 1],'linewidth',5) line([0 numel(dn120)+windowSize/2],[1 1],'LineStyle',':') title('GP120, Ka / Ks and epitopes'); ylabel('Ka / Ks'); xlabel('sliding window (middle codon)');
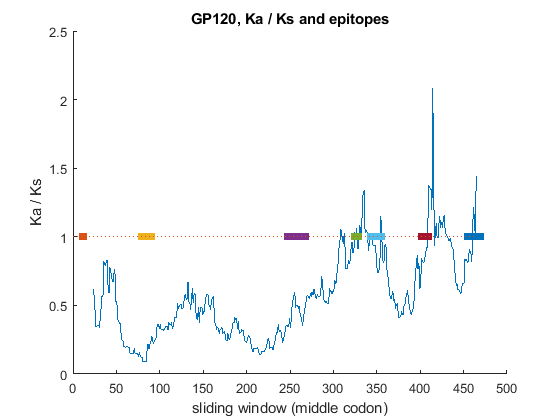
Although the general trend of the Ka/Ks ratio is less than 1, there are some regions where the ratio is greater than one, indicating that these regions are likely to be under positive selection. Interestingly, the location of some of these regions corresponds to the presence of T cell epitopes, identified by cellular methods. These segments display high amino acid variability because amino acid diversity in these regions allows the virus to evade the host immune system recognition. Thus, we can conclude that the source of variability in this regions is likely to be the host immune response.
References
[1] Cristianini, N. and Hahn, M.W., "Introduction to Computational Genomics: A Case Studies Approach", Cambridge University Press, 2007.
[2] Siebert, S.A., et al., "Natural Selection on the gag, pol, and env Genes of Human Immunodeficiency Virus 1 (HIV-1)", Molecular Biology and Evolution, 12(5):803-813, 1995.
You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)